

Next: Comparing Structures and Sequences
Up: VMD Tutorial
Previous: Data Analysis in VMD
Subsections
In this section you will learn to deal with multiple molecules within one VMD session. We will use the water transporting channel protein, aquaporin, as an example.
Aquaporins are membrane channel proteins found in a wide range of species, from bacteria to plants to human. They facilitate water transport across the cell membrane, and play an important role in the control of cell volume and transcellular water traffic. Many aquaporin protein structures are available in the Protein Data Bank, including the human aquaporin (PDB code 1FQY; Murata et al., Nature, 407:599, 2000) and E. coli aquaporin (PDB code 1RC2; Savage et al., PLoS Biology, 1:E72, 2003). To practice dealing with multiple proteins with VMD, let's load both aquaporin structures.
- 1
- Start a new VMD session. In the VMD Main window, choose File
 New Molecule.... The Molecule File Browser window should appear on your screen.
New Molecule.... The Molecule File Browser window should appear on your screen.
- 2
- Use the Browse... button to find the file 1fqy.pdb in vmd-tutorial-files in the tutorial directory. When you select the file, you will be back in the Molecule File Browser window. Press the Load button to load the molecule. The coordinate file of human aquaporin AQP1 should now be loaded and can be seen in the OpenGL window.
- 3
- The Molecule File Browser window should still be open; if not open it through File
New Molecule... again. Make sure you choose New Molecule in the Load files for: pull-down menu on the top. Use the Browse... button to find the file 1rc2.pdb in vmd-tutorial-files directory and press Load. Close the Molecule File Browser window.
You have just loaded a second molecule; any number of molecules may be loaded and displayed in VMD simultaneously by repeating the previous step. VMD can load as many molecules as the memory of your computer allows.
Take a look at your VMD Main window, which should look like Fig. 28. Within the VMD Main menu you can find the Molecule List Browser (circled in Fig. 28), which shows the global status of the loaded molecules. The Molecule List Browser displays information about each molecule, including Molecule ID (ID), the four Molecule Status Flags (T, A, D, and F, which stand for Top, Active, Drawn, and Fixed), name of the molecule (Molecule), number of atoms in the molecule (Atoms), number of frames loaded in the molecule (Frames), and the volumetric data loaded (Vol).
Figure 28:
The Molecule List Browser.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.7\textwidth]{FIGS/mol_list_browser}
}
\end{center}
\end{figure}](img77.gif) |
Changing molecule names
Let's first start with the Molecule column. By default the Molecule column displays file names of the molecules loaded in VMD, but you can change the molecule names to recognize them more easily.
- 4
- In the VMD Main menu, double-click on 1fqy.pdb in the Molecule column. A window will pop up with the message ``Enter a new name for molecule 0:" (Fig. 28a). Type in ``human aquaporin", and click OK (or press enter). In the VMD Main menu, the first molecule now has the name ``human aquaporin".
- 5
- Repeat the previous step for the E. coli aquaporin by double-clicking the 1rc2.pdb molecule name, and changing it to ``E. coli aquaporin" in the pop-up window. Your VMD Main window should now look like Fig. 29b.
Figure 29:
Changing molecule names.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.9\textwidth]{FIGS/change_mol_name}
}
\end{center}
\end{figure}](img78.gif) |
Before we continue exploring other features in the Molecule List Browser, take a look at your OpenGL Display window. You have two aquaporin structures, but since they are both shown in the same default representation, it is difficult to distinguish them. To tell them apart, you can assign them different representations.
- 6
- Open the Graphical Representations window via Graphics
Representations... from the VMD Main menu. Make sure 0:human aquaporin is selected in the Selected Molecule pull-down menu on top. Select New Cartoon for Drawing method, and ColorID
1 red for Coloring Method.
- 7
- In the Graphical Representations window, select 1:E. coli aquaporin in the Selected Molecule pull-down menu on top. Select New Cartoon for Drawing method, and ColorID
4 yellow for Coloring Method. Close the Graphical Representations window.
Now your OpenGL Display window should show a human aquaporin colored in red and an E. coli aquaporin colored in yellow (Fig. 30).
Figure 30:
The two aquaporins drawn in different representations.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.6\textwidth]{FIGS/two_aqp}
}
\end{center}
\end{figure}](img79.gif) |
In your OpenGL Display window, try moving the aquaporins around with your mouse in different mouse modes (rotating, scaling, and translating). You can see that both aquaporins move together. You can fix any molecule by double-clicking the F (fixed) flag in the Molecule List Browser on the left of the molecule name.
- 8
- In the Molecule List Browser, double-click on the F flag on the left of human aquaporin to fix the human aquaporin molecule. Return to the OpenGL Display window and toggle your mouse around. You can see that only the yellow E. coli aquaporin moves. Double-click on the F flag for human aquaporin again to release it.
One thing to notice about the F flag is that, although it may seem that one molecule has been moved relative to another when one of the molecules is fixed, the difference is only apparent. The internal coordinates of molecules are not changed by the rotation, translation and scaling motions. To change the coordinates of atoms in a molecule you need to use the text command interface (discussed in Section 3.2.3), and by using the atom move picking modes (by choosing Mouse
Move in the VMD Main menu).
Other features in the Molecule List Browser includes the Molecule ID (ID), Top (T), Active (A), and Drawn (D). Molecule ID is a number (starting from 0) assigned to each molecule when it's loaded into VMD, and is how VMD recognizes each molecule internally. You also refer to molecules by their Molecule IDs in text command interface. Top flag (T) indicates the default molecule in VMD operations, for example when resetting the VMD OpenGL view and when playing molecule trajectories. There can be only one top molecule at a time. Active flag (A) indicates if the trajectory of the given molecule is updated when using animation tools described in Section 2. Finally, Drawn flag (D) indicates if the given molecule is displayed in the OpenGL window. Let's try out the Top and Drawn flags.
- 9
- Make sure no molecule is fixed. By default the last molecule loaded in the VMD is the top molecule, so you can check and see that there is a T displayed for the E. coli aquaporin in the VMD Main menu. Reset the view by pressing ``
 " in the OpenGL Display window. Note that the yellow E. coli aquaporin is now placed in the center of the OpenGL Display window.
" in the OpenGL Display window. Note that the yellow E. coli aquaporin is now placed in the center of the OpenGL Display window.
- 10
- Switch the top molecule by double-clicking on the empty T flag for the human aquaporin molecule in the VMD Main menu. A T should appear for the human aquaporin, while the T for E. coli disappears. Go to the OpenGL Display window and reset the view again. You can see that this time the red human aquaporin is placed in the center of the OpenGL Display window.
- 11
- In the VMD Main menu, try hiding a molecule by double-clicking on its D flag. You can display the molecule again by double-clicking its D flag again.
When you look at your OpenGL Display window, you can see that the two aquaporins are very similar in structure. But it is difficult to detect their slight structural differences as the two proteins are placed apart. We will now try out a very useful Tcl command measure fit to align two molecules.
Figure 31:
Result of the alignment between the two aquaporins using the measure fit command.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.4\textwidth]{FIGS/measure_fit}
}
\end{center}
\end{figure}](img81.gif) |
- 1
- Open the VMD TkConsole window by choosing Extension
TkConsole from the VMD Main menu, and input the following commands (hit Enter after each line):
| set sel0 [atomselect 0 all] |
|
| set sel1 [atomselect 1 all] |
|
| set M [measure fit $sel0 $sel1] |
|
| $sel0 move $M |
|
![\framebox[0.9\textwidth]{
\par
\begin{tabular}{ll}
{\tt measure fit} {\it select...
... \\
coordinates of selection1 with the coordinates of selection2
\end{tabular}}](img82.gif)
As soon as you enter the last command line, you can see that the two aquaporins are now overlapping (Fig. 31). The 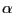 -helical regions of the aquaporins agree very well, with bigger deviations in the loop regions. Note the measure fit command can only work if two molecules have the same number of atoms. In this case it's a pure coincidence that the human aquaporin and E. coli aquaporin PDB files have the same number of atoms. The measure fit command is hence most useful in aligning the same protein in different conformations or different frames of a molecular dynamics simulation trajectory. Generally, to compare the structures of different proteins, one needs to use a different method. A good tool is the MultiSeq VMD plugin, which we will discuss in the following section.
-helical regions of the aquaporins agree very well, with bigger deviations in the loop regions. Note the measure fit command can only work if two molecules have the same number of atoms. In this case it's a pure coincidence that the human aquaporin and E. coli aquaporin PDB files have the same number of atoms. The measure fit command is hence most useful in aligning the same protein in different conformations or different frames of a molecular dynamics simulation trajectory. Generally, to compare the structures of different proteins, one needs to use a different method. A good tool is the MultiSeq VMD plugin, which we will discuss in the following section.
- 2
- Quit VMD.
Next: Comparing Structures and Sequences
Up: VMD Tutorial
Previous: Data Analysis in VMD
vmd@ks.uiuc.edu