

Next: Data Analysis in VMD
Up: VMD Tutorial
Previous: Trajectories and Movie Making
Subsections
VMD provides embedded scripting languages (Python and Tcl) for the purpose of user extensibility. In this section we will discuss the basic features of the Tcl scripting interface in VMD. You will see that everything you can do in VMD interactively can also be done with Tcl commands and scripts, and how the extensive list of Tcl text commands can help you investigate molecule properties and perform analysis.
To execute Tcl commands, you will be using a convenient text console called Tk Console.
- 1
- Start a new VMD session. In the VMD Main menu select Extensions
 Tk Console to open the VMD TkConsole window (Fig. 21). You can now start entering Tcl/Tk commands here.
Tk Console to open the VMD TkConsole window (Fig. 21). You can now start entering Tcl/Tk commands here.
Figure 21:
The VMD Tk Console window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/u4_tkcon}
}
\end{center}
\end{figure}](img46.gif) |
Let's start with the very basics of Tcl/Tk. Here are Tcl's set and puts commands:
- 2
- Try entering the following commands in the VMD TkConsole window. Remember to hit enter after each line and take a look at what you get after each input.
| set x 10 |
|
| puts "the value of x is:$x" |
|
| set text "some text" |
|
| puts "the value of text is:$text." |
|
As you can see, $variable refers to the value of variable.
Here is a command that performs mathematical operations:
- 3
- Try the expr command by entering the following lines in the VMD TkConsole window:
| expr 3 - 8 |
|
| set x 10 |
|
| expr - 3 * $x |
|
One of the most important aspects of Tcl is that you can embed Tcl commands into others by using brackets. A bracketed expression will automatically be substituted by the return value of the expression inside the brackets:
- 4
- Create some commands using brackets and test them. Try entering the following example in the VMD TkConsole window:
| set result [ expr -3 * $x ] |
|
| puts $result |
|
Often, one needs to execute a block of codes for many times. For this purpose, Tcl provides an iterated loop similar to the for loop in C. The for command in Tcl requires four arguments: an initialization, a test, an increment, and the block of code to evaluate. The syntax of the for command is:
- 5
- Now let's calculate the values of
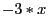 for integers
for integers 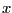 from 0 to 10 and output the results into a file named myoutput.dat. Please also pay attention the way of writing the output to a file on disk.
from 0 to 10 and output the results into a file named myoutput.dat. Please also pay attention the way of writing the output to a file on disk.
| set file [open "myoutput.dat" w] |
|
| for {set x 0} {$x <= 10} {incr x} { |
|
| puts $file [ expr -3 * $x ] |
|
| } |
|
| close $file |
|
- 6
- Take a look at the output file myoutput.dat, either by a text editor of your choice, or the command less in a terminal window on a Mac or Linux Machine.
Anything that can be done in the VMD graphical interface can be done with text commands. This allows scripts to be written that can automatically load molecules, create representations, analyze data, make movies, etc. Here, we will go through some simple examples of what can be done using the scripting interface in VMD.
- 1
- In the VMD TkConsole window, type the command mol new 1ubq.pdb and hit enter. As you can see, this command performs the same function as described at the beginning of Section 1.1, namely, loading a new molecule with file name 1ubq.pdb.
When you open VMD, by default a vmd console window appears. The vmd console window tells you what's going on within the VMD session that you are working on.
- 2
- Take a look at the vmd console window (Fig. 22). It should tell you a molecule has been loaded, as well as some of its basic properties like number of atoms, bonds, residues and etc. The Tcl commands that you enter in the VMD TkConsole window can also be entered in the vmd console window. If you are using a Mac, your vmd console window is the terminal window that shows up when you open VMD.
Figure 22:
The VMD Console window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.9\textwidth]{FIGS/u4_vmdcon}
}
\end{center}
\end{figure}](img54.gif) |
Many times you might want to perform operations on only a specific part a molecule. For this purpose, VMD's atomselect command is very useful.
This command allows you to select a specific part of a molecule. The first argument to atomselect is the molecule ID (shown to the very left of the VMD Main window), the second argument is a textual atom selection like what you have been using to describe graphical representations in Section 1.3. The selection returned by atomselect is itself a command which you will learn to use.
- 3
- Type set crystal [atomselect top "all"] in the Tk Console window. This creates a selection, crystal, that contains all the atoms in the molecule and assigns it to the variable crystal. Instead of a molecule ID (which is a number), we have used the shortcut ``top" to refer to the top molecule. A top molecules means that it is the target for scripting commands. This concept is particularly important when multiple molecules are loaded at the same time (see Section 4 for dealing with multiple molecules in VMD).
The result of atomselect is a function. Thus, $crystal is now a function that performs actions on the contents of the ``all" selection.
Obtaining and changing molecule properties with text commands
After you have defined an atom selection, you have many commands
that you can use to operate on it. For example, you can use commands to
learn about the properties (number of atoms, coordinates, total charge, etc)
of your atom selection. You can also use commands to change its coordinates
and other properties.
See VMD User's Guide2for an extensive list of
commands.
- 4
- Type $crystal num in the Tk Console window. Passing num to an atom selection returns the number of atoms in that selection. Check that this number matches the number of atoms for your molecule displayed in the VMD Main window.
- 5
- We can also use commands to move our molecule on the screen. You can use these commands to change atom coordinates.
| $crystal moveby {10 0 0} |
|
| $crystal move [transaxis x 40 deg] |
|
The following examples will show you how to edit atomic properties using VMD's atomselect command.
- 6
- Open the Graphical Representation window by selecting Graphics
Representations... in the VMD Main window. Type in protein as the atom selection, change its Coloring Method to Beta and its Drawing Method to VDW. Your molecule should now appear as a mostly red and blue assembly of spheres.
- 7
- Return to the Tk Console window, and type $crystal set beta 0. This resets the ``beta" field (which is displayed) to zero for all atoms. As you do this, you should observe that the atoms in your OpenGL window will suddenly change to a uniform color (since they all have the same beta values now).
Atom selections are just references to the atoms in the original molecule. When you change a property (e.g. beta value) of some atoms through a selection, that change is reflected in all the other selections that contain those atoms.
- 8
- In the Tk Console window, type set sel [atomselect top "hydrophobic"]. This creates a selection, sel, that contains all the atoms in the hydrophobic residues.
- 9
- Let's label all hydrophobic atoms by setting their beta values to 1. You probably know how to do this by now: type $sel set beta 1 in the Tk Console window. If the colors in the OpenGL Display do not get updated, go to the Graphical Representations window and click on the Apply button at the bottom.
- 10
- You will now change a physical property of the atoms to further illustrate the distribution of hydrophobic residues. In the Tk Console window type $crystal set radius 1.0 to make all the atoms smaller and easier to see through, and then $sel set radius 1.5 to make atoms in the hydrophobic residues larger. The radius field affects the way that some representations (e.g., VDW, CPK) are drawn.
You have now created a visual state that clearly distinguishes which parts of the protein are hydrophobic and which are hydrophilic. If you have followed the instructions correctly, your protein should resemble Fig. 23.
Figure 23:
Ubiquitin in the VDW representation, colored according to the hydrophobicity of its residues.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/u4_hydrophobicity}
}
\end{center}
\end{figure}](img58.gif) |
Atom selections are useful not only for setting atomic data, but also for getting atomic information. Let's say that you wish to communicate which residues are hydrophobic, all you need to do is to create a hydrophobic selection and use get command.
- 11
- Try to use get command with your sel atom selection to obtain the names of hydrophobic residues:
But there is a problem! Each residue contains many atoms, resulting in multiple repeated entries. Can you think of a way to circumvent this? We know that each amino acid has the same backbone atoms. If you pick only one of these atoms per residue, each residue will be present only once in your selection.
- 12
- Let's try this solution. Each residue has one and only one
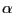 -carbon (name CA = alpha), so type the following in the Tk Console window:
-carbon (name CA = alpha), so type the following in the Tk Console window:
| set sel [atomselect top "hydrophobic and alpha"] |
|
| $sel get resname |
|
This should give you the list of hydrophobic residues.
- 13
- You can also get multiple properties simultaneously. Try the following:
| $sel get resid |
|
| $sel get {resname resid} |
|
| $sel get {x y z} |
|
If you want to obtain some of the structural properties, e.g., the geometric center or the size of a selection, the command measure can do the job easily.
- 14
- Let's try using measure with the sel selection:
| measure center $sel |
|
| measure minmax $sel |
|
The first command above returns the geometric center of atoms in sel. And the second command returns two vectors, the first containing the minimum
, 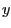 , and
, and  coordinates of all atoms in sel, and the second containing the corresponding maxima.
coordinates of all atoms in sel, and the second containing the corresponding maxima.
- 15
- Once you are done with a selection, it is always a good ideal to delete it to save memory:
We have learned many useful commands in VMD. When performing a task that requires many lines of commands, instead of typing each line in the Tk Console window, it is usually more convenient to write all the lines into a script file and load it in VMD. This is very easy to do. Just use any text editor to write your script file, and in a VMD session, use the command source filename to execute the file.
In the vmd-tutorial-file, you will find a simple script file beta.tcl, which we will execute in VMD as an example. The script beta.tcl sets the colors of residues LYS and GLY to a different color from the rest of the protein by assigning them a different beta value, a trick you have also learned in Section 3.2.3.
- 16
- In the Tk Console window, type source beta.tcl and observe the color change. You should see that the protein is mostly a collection of red spheres, with some residues shown in blue. The blue residues are the LYS and GLY residues in the ubiquitin.
- 17
- Let's take a quick look at the beta.tcl. Use any text editor of your choice, open the file beta.tcl. You can see that there are six lines in this file, and each line represents a Tcl command line that you have used before. Close the text editor when you are done.
VMD offers a way to display user-defined objects built from graphics primitives such as points, lines, cylinders, cones, spheres, triangles, and text.
The command that can realize those functions is graphics, the syntax of which is
Where molid is a valid molecule ID and command is one of the commands shown below. Let's try drawing some shapes with the following examples.
- 1
- Hide all representations in the Graphical Representations window.
- 2
- Let's draw a point. Type the following command in your Tk Console window:
| graphics top point {0 0 10} |
|
Somewhere in your OpenGL window there should be a small dot.
- 3
- Let's draw a line. Type the following command in your Tk Console window:
| graphics top line {-10 0 0} {0 0 0} width 5 style solid |
|
This will give you a solid line.
- 4
- You can also draw a dashed line:
| graphics top line {10 0 0} {0 0 0} width 5 style dashed |
|
- 5
- All the objects drawn so far are all in blue. You can change the color of the next graphics object by using the command graphics top color colorid. The colorid for each color can be found in Graphics
Colors... menu in VMD Main window. For example, the color for orange is ``3". Type graphics top color 3 in the Tk Console window, and the next object you draw will appear in orange.
- 6
- Try the following commands to draw more shapes:
| graphics top cylinder {15 0 0} {15 0 10} radius 10 resolution 60 filled no |
|
| graphics top cylinder {0 0 0} {-15 0 10} radius 5 resolution 60 filled yes |
|
| graphics top cone {40 0 0} {40 0 10} radius 10 resolution 60 |
|
| graphics top sphere {65 0 5} radius 10 resolution 80 |
|
| graphics top triangle {80 0 0} {85 0 10} {90 0 0} |
|
| graphics top text {40 0 20} "my drawing objects" |
|
- 7
- On your OpenGL window, there are a lot of objects now. To find the list of objects you've drawn, use the command graphics top list. You'll get a list of numbers, standing for the ID of each object.
- 8
- The detailed information about each object can be obtained by typing graphics top info ID. For example, type graphics top info 0 to see the information on the point you drew.
- 9
- You can also delete some of the unwanted objects using the command graphics top delete ID .
- 10
- Using these basic shape-drawing commands, you can create geometric objects, as well as texts, to be displayed in your OpenGL window. When you render an image (as discussed in Section 1.6.5), these objects will be included in the resulting image file. You can hence use geometric objects and texts to point or label interesting features in your molecule. When you are done, quit VMD.
Next: Data Analysis in VMD
Up: VMD Tutorial
Previous: Trajectories and Movie Making
vmd@ks.uiuc.edu
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...laddnormallink{http://www.tcl.tk/doc}{http://www.tcl.tk/doc/}. }
\end{minipage}}](img45.gif)
![\framebox[0.9\textwidth]{
\par
\begin{tabular}{ll}
{\tt set} {\it variable} {\it...
...\tt \$}{\it variable} & -- prints out the value of {\it variable}
\end{tabular}}](img47.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...dow to move to the correct directory where 1ubq.pdb is located.}
\end{minipage}}](img53.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...u want to show a property of the system that you have computed.}
\end{minipage}}](img56.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...and z), charge, mass, occupancy and radius, just to name a few.}
\end{minipage}}](img57.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...the protein achieve proper folding and increases its stability.}
\end{minipage}}](img59.gif)
![% latex2html id marker 5798
\framebox[\textwidth]{
\begin{minipage}{.2\textwidt...
...yping {\tt source myfirststate.vmd} in the {Tk Console} window.}
\end{minipage}}](img62.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...med interactively. To turn off logfile, type {\tt logfile off}.}
\end{minipage}}](img64.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...mation on the VMD startup files, refer to the VMD
user's guide.}
\end{minipage}}](img65.gif)