Next: Solute Scaling and REST2
Up: Accelerated Sampling Methods
Previous: Accelerated Molecular Dynamics
Contents
Index
Subsections
Gaussian Accelerated Molecular Dynamics
Gaussian accelerated molecular dynamics (GaMD) [76] is a type of accelerated molecular dynamics (aMD) calculation. It is an enhanced sampling method that works by adding a harmonic boost potential to smoothen the system's potential energy surface.
By constructing a boost potential that follows Gaussian distribution, accurate reweighting of the GaMD simulations is achieved using cumulant expansion to the second order.
Please include the following two references in your work using the NAMD implementation of GaMD:
- Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation, Y.Miao, V.Feher, and J.A. McCammon. J. Chem. Theory Comput., 11:3584-3595, 2015.
- Gaussian Accelerated Molecular Dynamics in NAMD, Y.T.Pang, Y.Miao, Y.Wang, and J.A. McCammon, J. Chem. Theory Comput., 13:9-19, 2017.
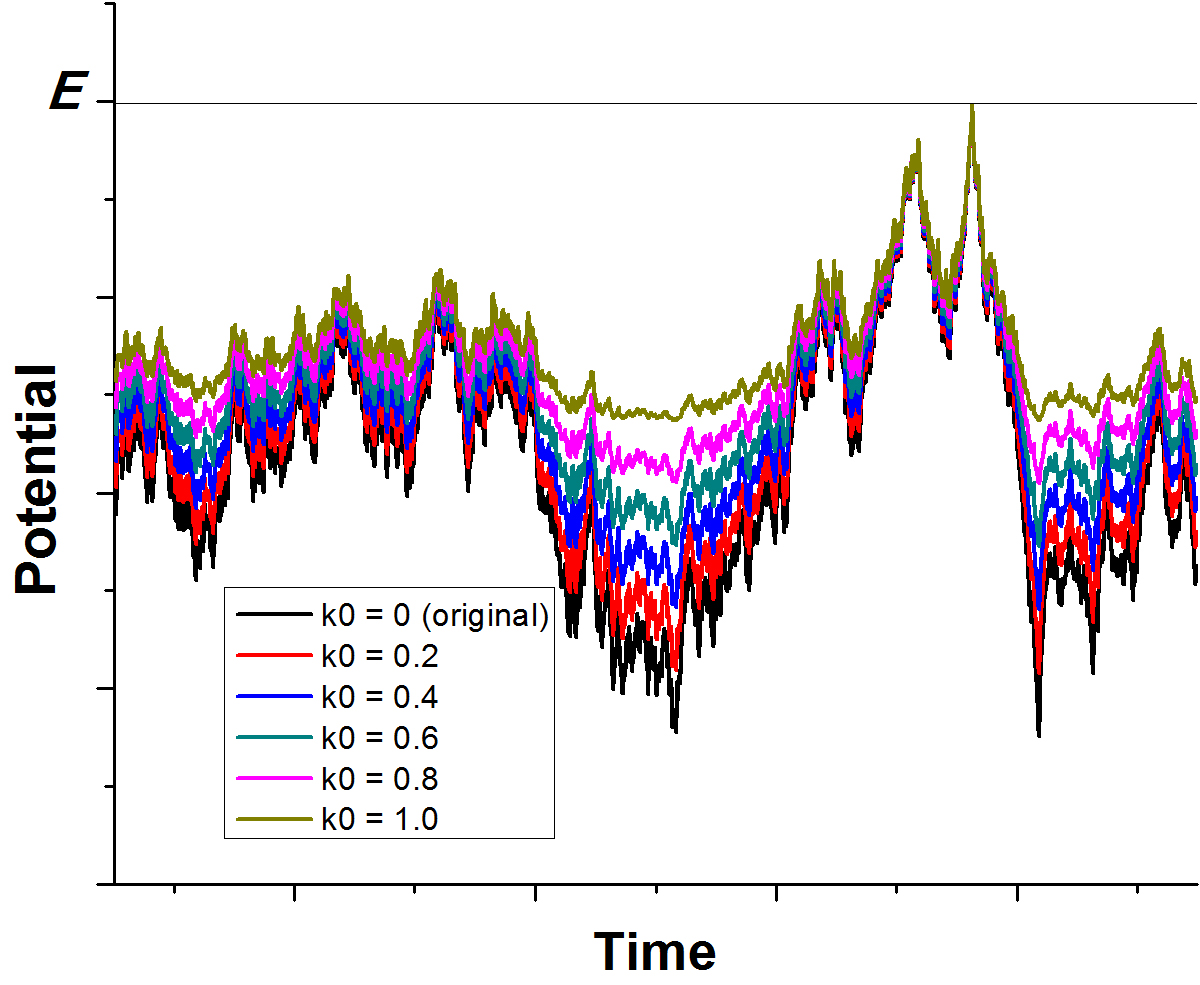
GaMD enhances conformational sampling of biomolecules by adding a harmonic boost potential to smoothen the system's potential energy surface [76], as illustrated below:
Figure:
Schematic illustration of GaMD. When the threshold energy 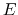 is set to the maximum potential (
is set to the maximum potential (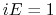 mode), the system's potential energy surface is smoothened by adding a harmonic boost potential that follows a Gaussian distribution. The coefficient
mode), the system's potential energy surface is smoothened by adding a harmonic boost potential that follows a Gaussian distribution. The coefficient 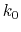 , which falls in the range of
, which falls in the range of 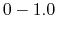 , determines the magnitude of the applied boost potential.
, determines the magnitude of the applied boost potential.
|
|
Consider a system with 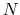 atoms at positions
atoms at positions
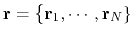 .
When the system's potential energy
.
When the system's potential energy
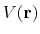 is lower than a threshold energy
, the following boost potential is added:
is lower than a threshold energy
, the following boost potential is added:
 |
(85) |
where
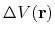 is the boost potential,
is the boost potential,
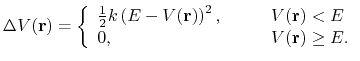 |
(86) |
where 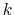 is the harmonic force constant.
is the harmonic force constant.
As explained in reference [76], the two adjustable parameters
and
are automatically determined by the following three criteria.
First, 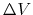 should not change the relative order of the biased potential values, i.e., for any two arbitrary potential values
should not change the relative order of the biased potential values, i.e., for any two arbitrary potential values
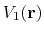 and
and
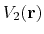 found on the original energy surface, if
found on the original energy surface, if
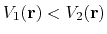 ,
then one should have
,
then one should have
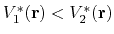 .
Second, the difference between potential energy values on the smoothened energy surface should be smaller than that of the original,
i.e., if
, then one should have
.
Second, the difference between potential energy values on the smoothened energy surface should be smaller than that of the original,
i.e., if
, then one should have
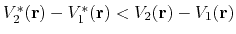 .
By combining the above two criteria and plugging in the formula of
.
By combining the above two criteria and plugging in the formula of
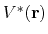 and
, one obtains
and
, one obtains
where
 min
and
max
are the system's minimum and maximum potential energies. To ensure that Eqn.(88) is valid,
needs
to satisfy:
min
and
max
are the system's minimum and maximum potential energies. To ensure that Eqn.(88) is valid,
needs
to satisfy:
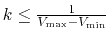 .
Define
.
Define
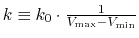 , then
, then
 .
Third, the standard deviation of
needs to be small enough (i.e., narrow distribution) to ensure accurate reweighting using cumulant expansion to the second order:
.
Third, the standard deviation of
needs to be small enough (i.e., narrow distribution) to ensure accurate reweighting using cumulant expansion to the second order:
 ,
where
avg
and
,
where
avg
and  are the average and standard deviation of the system's potential energies,
are the average and standard deviation of the system's potential energies,
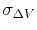 is the standard deviation of
, while
is the standard deviation of
, while 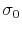 is a user-specified upper limit (e.g.,
is a user-specified upper limit (e.g., 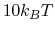 ) in order to achieve accurate reweighting.
) in order to achieve accurate reweighting.
iE = 1 mode: When
is set to
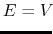 max
according to Eqn.(88),
is calculated as:
max
according to Eqn.(88),
is calculated as:
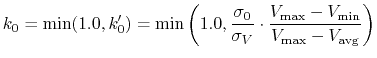 |
(88) |
iE = 2 mode: Alternatively, when
is set to
min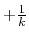 ,
is calculated as:
,
is calculated as:
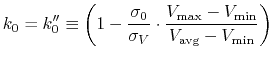 |
(89) |
If 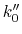 obtained from the above equation is smaller than 0 or greater than 1, then
will be calculated using Eqn.(89).
obtained from the above equation is smaller than 0 or greater than 1, then
will be calculated using Eqn.(89).
For more details on GaMD and the corresponding reweighting using cumulant expansion, see reference [76][85].
Same as aMD, three modes are available for applying boost potential in GaMD:
(1) boosting the dihedral energy only,
(2) boosting the total potential energy, and
(3) boosting both the dihedral and total potential energy (i.e., ``dual-boost").
Some parameters from aMD, including: accelMD, accelMDdihe, accelMDdual, accelMDFirstStep, accelMDLastStep and accelMDOutFreq are shared by GaMD (see Section 11.1 for details).
The following is a list of input parameters unique to a GaMD run:
- accelMDG
 Is Gaussian accelerated MD on?
Is Gaussian accelerated MD on? 
Acceptable Values: on or off
Default Value: off
Description: Specifies whether Gaussian accelerated MD (GaMD) is on. Only available when accelMD is on.
- accelMDGiE
Flag to set the threshold energy for adding boost potential
Acceptable Values: 1 or 2
Default Value: 1
Description: Specifies how the threshold energy
is set in GaMD. A value of 1 indicates that the threshold energy
is set to its lower bound
max
. A value of 2 indicates that the threshold energy is set to its upper bound
min max
max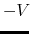 min
min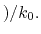
- accelMDGcMDPrepSteps
Number of preparatory cMD steps
Acceptable Values: Zero or Positive integer
Default Value: 200,000
Description: The number of preparatory conventional MD (cMD) steps in GaMD. This value should be smaller than accelMDGcMDSteps (see below). Potential energies are not collected for calculating the values of
max
,
min
,
avg
,
during the first accelMDGcMDPrepSteps.
- accelMDGcMDSteps
Number of total cMD steps
Acceptable Values: Zero or Positive integer
Default Value: 1,000,000
Description: The number of total cMD steps in GaMD. With
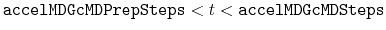 ,
max
,
min
,
avg
,
are collected and at
,
max
,
min
,
avg
,
are collected and at
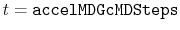 ,
and
are computed.
,
and
are computed.
- accelMDGEquiPrepSteps
Number of preparatory equilibration steps in GaMD
Acceptable Values: Zero or Positive integer
Default Value: 200,000
Description: The number of preparatory equilibration steps in GaMD. This value should be smaller than accelMDGEquiSteps (see below). With
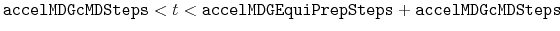 , GaMD boost potential is applied according to
and
obtained at
.
, GaMD boost potential is applied according to
and
obtained at
.
- accelMDGEquiSteps
Number of total equilibration steps in GaMD
Acceptable Values: Zero or Positive integer
Default Value: 1,000,000
Description: The number of total equilibration steps in GaMD. With
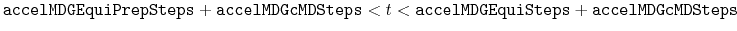 , GaMD boost potential is applied, and
and
are updated every step.
, GaMD boost potential is applied, and
and
are updated every step.
- accelMDGStatWindow
Number of steps to calculate average and standard deviation in GaMD
Acceptable Values: Integer
Default Value: -1
Description: The number of simulation steps used to calculate the average and standard deviation of potential energies, as well as the frequency of recalculating the boost potential during equilibration steps. When it is set to a negative number, all the steps throughout the cMD and equilibration stage (except the preparatory steps) will be used to calculate the average and standard deviation without resetting, and the boost potential will be updated every step during equilibration steps. When used, it is recommended to be set to about 4 times the total number of atoms in the system. Note that accelMDGcMDPrepSteps, accelMDGcMDSteps, accelMDGEquiPrepSteps and accelMDGEquiSteps need to be multiples of accelMDGStatWindow.
- accelMDGSigma0P
Upper limit of the standard deviation of the total boost potential in GaMD
Acceptable Values: Positive real number
Default Value: 6.0 (kcal/mol)
Description: Specifies the upper limit of the standard deviation of the total boost potential. This option is only available when accelMDdihe is off or when accelMDdual is on.
- accelMDGSigma0D
Upper limit of the standard deviation of the dihedral boost potential in GaMD
Acceptable Values: Positive real number
Default Value: 6.0 (kcal/mol)
Description: Specifies the upper limit of the standard deviation of the dihedral boost potential. This option is only available when accelMDdihe or accelMDdual is on.
- accelMDGRestart
Flag to restart GaMD simulation
Acceptable Values: on or off
Default Value: off
Description: Specifies whether the current GaMD simulation is the continuation of a previous run. If this option is turned on, the GaMD restart file specified by accelMDGRestartFile (see below) will be read.
- accelMDGRestartFile
Name of GaMD restart file
Acceptable Values: UNIX filename
Description: A GaMD restart file that stores the current number of steps, maximum, minimum, average and standard deviation of the dihedral and/or total potential energies (depending on the accelMDdihe and accelMDdual parameters). This file is saved automatically every restartfreq steps. If accelMDGRestart is turned on, this file will be read and the simulation will restart from the point where the file was written.
Next: Solute Scaling and REST2
Up: Accelerated Sampling Methods
Previous: Accelerated Molecular Dynamics
Contents
Index
http://www.ks.uiuc.edu/Research/namd/