



Next: Tcl Boundary Forces
Up: User Defined Forces
Previous: Interactive Molecular Dynamics (IMD)
Contents
Index
NAMD provides a limited Tcl scripting interface designed for applying forces and performing on-the-fly analysis.
This interface is efficient if only a few coordinates, either of individual atoms or centers of mass of groups of atoms, are needed.
In addition, information must be requested one timestep in advance.
To apply forces individually to a potentially large number of atoms, use
tclBC instead as described in Sec. 9.11.
The following configuration parameters are used to enable the Tcl interface:
At this point only low-level commands are defined.
In the future this list will be expanded. Current commands are:
- print <anything>
This command should be used instead of puts to display output.
For example, ``print Hello World''.
- atomid <segname> <resid> <atomname>
Determines atomid of an atom from its segment, residue, and name.
For example, ``atomid br 2 N''.
- addatom <atomid>
Request coordinates of this atom for next force evaluation, and
the calculated total force on this atom for current force evaluation.
Request remains in effect until clearconfig is called.
For example, ``addatom 4'' or ``addatom [atomid br 2 N]''.
- addgroup <atomid list>
Request center of mass coordinates of this group for next force evaluation.
Returns a group ID which is of the form gN where N is a small integer.
This group ID may then be used to find coordinates and apply forces just like a regular atom ID.
Aggregate forces may then be applied to the group as whole.
Request remains in effect until clearconfig is called.
For example, ``set groupid [addgroup { 14 10 12 }]''.
- clearconfig
Clears the current list of requested atoms. After clearconfig,
calls to addatom and addgroup can be used to build a new
configuration.
- getstep
Returns the current step number.
- loadcoords <varname>
Loads requested atom and group coordinates (in Å) into a local array.
loadcoords should only be called from within the calcforces procedure.
For example, ``loadcoords p'' and ``print $p(4)''.
- loadforces <varname>
Loads the forces applied in the previous timestep (in kcal mol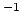 Å
) into a local array.
loadforces should only be called from within the calcforces procedure.
For example, ``loadforces f'' and ``print $f(4)''.
Å
) into a local array.
loadforces should only be called from within the calcforces procedure.
For example, ``loadforces f'' and ``print $f(4)''.
- loadtotalforces <varname>
Loads the total forces on each requested atom in the previous
time step (in kcal mol
Å
) into a local array. The
total force also includes external forces. Note that the
``loadforces'' command returns external forces applied by
the user. Therefore, one can subtract the external force on an
atom from the total force on this atom to get the pure force
arising from the simulation system.
- loadmasses <varname>
Loads requested atom and group masses (in amu) into a local array.
loadmasses should only be called from within the calcforces procedure.
For example, ``loadcoords m'' and ``print $m(4)''.
- addforce <atomid|groupid> <force vector>
Applies force (in kcal mol
Å
) to atom or group.
addforce should only be called from within the calcforces procedure.
For example, ``addforce $groupid { 1. 0. 2. }''.
- addenergy <energy (kcal/mol)>
This command adds the specified energy to the MISC column (and
hence the total energy) in the energy output. For normal runs,
the command does not affect the simulation trajectory at all, and
only has an artificial effect on its energy output. However, it
can indeed affect minimizations.
With the commands above and the functionality of the Tcl language,
one should be able to perform any on-the-fly analysis and
manipulation. To make it easier to perform certain tasks,
some Tcl routines are provided below.
Several vector routines (vecadd, vecsub, vecscale)
from the VMD Tcl interface are defined. Please refer to VMD
manual for their usage.
The following routines take atom coordinates as input, and return
some geometry parameters (bond, angle, dihedral).
- getbond <coor1> <coor2>
Returns the length of the bond between the two atoms. Actually
the return value is simply the distance between the two
coordinates. ``coor1'' and ``coor2'' are coordinates of the atoms.
- getangle <coor1> <coor2> <coor3>
Returns the angle (from 0 to 180) defined by the
three atoms. ``coor1'', ``coor2'' and ``coor3'' are coordinates of
the atoms.
- getdihedral <coor1> <coor2> <coor3> <coor4>
Returns the dihedral (from -180 to 180) defined by the
four atoms. ``coor1'', ``coor2'', ``coor3'' and ``coor4'' are
coordinates of the atoms.
The following routines calculate the derivatives (gradients) of
some geometry parameters (angle, dihedral).
As an example, here's a script which applies a harmonic
constraint (reference position being 0) to a dihedral. Note that
the ``addenergy'' line is not really necessary - it simply adds
the calculated constraining energy to the MISC column, which is
displayed in the energy output.
tclForcesScript {
# The IDs of the four atoms defining the dihedral
set aid1 112
set aid2 123
set aid3 117
set aid4 115
# The "spring constant" for the harmonic constraint
set k 3.0
addatom $aid1
addatom $aid2
addatom $aid3
addatom $aid4
set PI 3.1416
proc calcforces {} {
global aid1 aid2 aid3 aid4 k PI
loadcoords p
# Calculate the current dihedral
set phi [getdihedral $p($aid1) $p($aid2) $p($aid3) $p($aid4)]
# Change to radian
set phi [expr $phi*$PI/180]
# (optional) Add this constraining energy to "MISC" in the energy output
addenergy [expr $k*$phi*$phi/2.0]
# Calculate the "force" along the dihedral according to the harmonic constraint
set force [expr -$k*$phi]
# Calculate the gradients
foreach {g1 g2 g3 g4} [dihedralgrad $p($aid1) $p($aid2) $p($aid3) $p($aid4)] {}
# The force to be applied on each atom is proportional to its
# corresponding gradient
addforce $aid1 [vecscale $g1 $force]
addforce $aid2 [vecscale $g2 $force]
addforce $aid3 [vecscale $g3 $force]
addforce $aid4 [vecscale $g4 $force]
}
}
Next: Tcl Boundary Forces
Up: User Defined Forces
Previous: Interactive Molecular Dynamics (IMD)
Contents
Index
http://www.ks.uiuc.edu/Research/namd/