



Next: Steered Molecular Dynamics (SMD)
Up: User Defined Forces
Previous: Symmetry Restraints
Contents
Index
In TMD, subset of atoms in the simulation is guided towards a
final 'target' structure by means of steering forces. At each timestep,
the RMS distance between
the current coordinates and the target structure is computed (after
first aligning the target structure to the current coordinates).
The force on each atom is given by the gradient of the potential
![$\displaystyle U_{TMD} = \frac{1}{2} \frac{k}{N} \left[ RMS(t) - RMS^*(t) \right]^2$](img208.png) |
(31) |
where 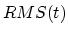 is the instantaneous best-fit RMS distance of the current
coordinates from the target coordinates, and
is the instantaneous best-fit RMS distance of the current
coordinates from the target coordinates, and 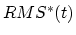 evolves linearly
from the initial RMSD at the first TMD step to the final RMSD at the last
TMD step. The spring constant
evolves linearly
from the initial RMSD at the first TMD step to the final RMSD at the last
TMD step. The spring constant 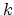 is scaled down by the number
is scaled down by the number 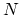 of targeted
atoms. Atoms can be separated into non-overlapping constraint domains by
assigning integer values in the beta column of the .pdb file. Forces on
the atoms will be calculated for each domain independently of the other domains.
of targeted
atoms. Atoms can be separated into non-overlapping constraint domains by
assigning integer values in the beta column of the .pdb file. Forces on
the atoms will be calculated for each domain independently of the other domains.
- TMD
 Is TMD active
Is TMD active 
Acceptable Values: on or off
Default Value: off
Description: Should TMD steering forces be applied to the system. If TMD is enabled,
TMDk, TMDFile, and TMDLastStep must be defined in the
input file as well.
- TMDk Elastic constant for TMD forces
Acceptable Values: Positive value in 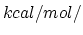 Å
Å .
.
Description: The value of in Eq. 32. A value of 200 seems to work
well in many cases. If this setting is omitted, the value in the occupancy column
of the pdb file specified by TMDFile will be used as the constant for that atom.
This allows the user to specify the constant on a per-atom basis.
- TMDOutputFreq How often to print TMD output
Acceptable Values: Positive integer
Default Value: 1
Description: TMD output consists of lines of the form TMD ts targetRMS currentRMS
where ts is the timestep, targetRMS is the target RMSD at that
timestep, and currentRMS is the actual RMSD.
- TMDFile File for TMD information
Acceptable Values: Path to PDB file
Description:
Target atoms are those whose occupancy (O) is nonzero in the TMD PDB file.
The file must contain the same number of atoms as the structure file. The
coordinates for the target structure are also taken from the targeted
atoms in this file. Non-targeted atoms are ignored. The beta column of targetted
atoms is used to designate non-overlapping constraint domains. Forces will be
calculated for atoms within a domain separately from atoms of other domains.
- TMDFirstStep first TMD timestep
Acceptable Values: Positive integer
Default Value: 0
Description:
- TMDLastStep last TMD timestep
Acceptable Values: Positive integer
Description: TMD forces are applied only between TMDFirstStep and TMDLastStep.
The target RMSD evolves linearly in time from the initial to the final target
value.
- TMDInitialRMSD target RMSD at first TMD step
Acceptable Values: Non-negative value in Å
Default Value: from coordinates
Description:
In order to perform TMD calculations that involve restarting a previous
NAMD run, be sure to specify TMDInitialRMSD with the same value
in each NAMD input file, and use the NAMD parameter firstTimestep
in the continuation runs so that the target RMSD continues from where the
last run left off.
- TMDFinalRMSD target RMSD at last TMD step
Acceptable Values: Non-negative value in Å
Default Value: 0
Description: If no TMDInitialRMSD is given, the initial RMSD will be calculated at the
first TMD step. TMDFinalRMSD may be less than or greater than
TMDInitialRMSD, depending on whether the system is to be steered
towards or away from a target structure, respectively. Forces are applied
only if is betwween TMDInitialRMSD and 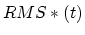 ; in other
words, only if the current RMSD fails to keep pace with the target value.
; in other
words, only if the current RMSD fails to keep pace with the target value.
- TMDDiffRMSD Is double-sided TMD active?
Acceptable Values: on or off
Default Value: off
Description: Turns on the double-sided TMD feature which targets the transition between two structures.
This is accomplished by modifying the TMD force such that the potential is based on the
difference of RMSD's from the two structures:
![$\displaystyle U_{TMD} = \frac{1}{2} \frac{k}{N} \left[ DRMS(t) - DRMS^*(t) \right]^2$](img214.png) |
(32) |
where 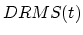 is RMS1(t) - RMS2(2) (RMS1 being the RMSD from structure 1 and RMS2 the RMSD from structure 2).
The first structure is specified as normal in TMDFile and the second structure should
be specified in TMDFile2, preserving any domain designations found in TMDFile.
is RMS1(t) - RMS2(2) (RMS1 being the RMSD from structure 1 and RMS2 the RMSD from structure 2).
The first structure is specified as normal in TMDFile and the second structure should
be specified in TMDFile2, preserving any domain designations found in TMDFile.
- TMDFile2 Second structure file for double-sided TMD
Acceptable Values: Path to PDB file
Description: PDB file defining the second structure of a double sided TMD. This file
should contain the same number of atoms as TMDFile along with the
same domain designations if any are specified.
Next: Steered Molecular Dynamics (SMD)
Up: User Defined Forces
Previous: Symmetry Restraints
Contents
Index
namd@ks.uiuc.edu